Electrostatic Tools Manual
Oleg I. Titov and Dmitry A. Shulga
22nd February 2015
Contents
1 About
Electrostatic Tools is a program package that aims to assist in developing of
next-generation molecular electrostatic models with an enhanced molecular
electrostatic potential (MEP) anisotropy. It provides the researcher with tools
to fit and for work with customizable atomic multipole moments (up to a
quadrupole) and extra-point charges. The models obtained are intended to use for
description of an electrostatic part of such highly anisotropic moieties as
oxygens, nitrogens with lone pairs and heavy halogens (Cl, Br, I) for halogen
bonding.
2 Installation
The basic installation requires a functioning C++ compiler and development
versions of all prerequisites installed. Electrostatic Tools currently depends
on:
- CMake is used to control the build and installation process
- OpenBabel library is used to read and write molecules in common
chemical formats and perform SMARTS pattern matching
- Eigen3 library provides linear algebra solvers
- boost is also required (with program_options binaries compiled)
- optional SWIG to build script bindings. Currently only Perl and Python
are supported
Common procedure for all cmake-based packages can be applied:
tar -xf electrostatic-tools-0.1.0.tar.bz2 cd electrostatic-tools-0.1.0 mkdir build cd build cmake .. make make install
One can be interested in the following options that can be passed to cmake:
- -DCMAKE_INSTALL_PREFIX=__prefix__ This tells cmake to install the
package in the __prefix__ directory. Most would like to use something
like ~/el_tools. The default is /usr/local.
- -DCMAKE_BUILD_TYPE=Release This will change build configuration to a
Release version meaning fast optimized executables. The other useful
option is RelWithDebInfo meaning the release-optimized executables with
a debugging info.
- -DET_NO_mult_fitter=ON/OFF, -DET_NO_ep_fitter=ON/OFF
and -DET_NO_mmol2mol=ON/OFF These switches turn off compilation and
installation of the corresponding programs. The defaults are OFF
- -DET_PERL_BINDINGS=ON/OFF and -DET_PYTHON_BINDINGS=ON/OFF
These switches turn off compilation and installation of the corresponding
language bindings.
- -DET_PERL_LIBDIR and -DET_PYTHON_LIBDIR Manually specify the
destination of language bindings.
- -DET_PERL_INCLUDE_PATH, -DET_PERL_LIBRARY,
-DET_PYTHON_INCLUDE_PATH and -DET_PYTHON_LIBRARY can be used to
override cmake’s guesses.
For example if you want to install the release-optimized package in your home
directory, you don’t need Perl bindings and you want to install Python bindings in
subfolder in your home directory, you should call:
cmake -DCMAKE_BUILD_TYPE=Release \ -DCMAKE_INSTALL_PREFIX=~/elec_tools \ -DET_PERL_BINDINGS=OFF \ -DET_PYTHON_LIBDIR=~/elec_tools/lib \ ..
To temporarly add Electrostatic Tools to a $PATH variable invoke:
export PATH=$PATH:__prefix__/bin
for bash shell, where __prefix__ is your installation prefix, or for csh shell:
setenv PATH $PATH:__prefix__/bin
To permanently add Electrostatic Tools to $PATH invoke:
echo "export PATH=$PATH:__prefix__/bin" >> ~/.bashrc
for bash shell, where __prefix__ is your installation prefix, or for csh shell:
echo "setenv PATH $PATH:__prefix__/bin" >> ~/.cshrc
3 Tutorial
In this section we will discuss the usage of our programs. Files used in this
tutorial are shipped with the source package in an examples directory. We
assume that the package is compiled, installed and added to $PATH. Every
program in Electrostatic Tools has a build-in help available with a --help
switch.
3.1 Multipole Fit
To perform a multipole fit you will need a molecule structure in any common file
formats supported by OpenBabel and a reference electrostatic potential in .esp
format (used for a RESP-charges fitting within AmberTools package). The
sample files with a phenylbromide molecule (phbr.mol2) and the reference
RHF/6-31G* potential (phbr.esp) are located in the examples directory of
the source tree. We’ll make a temporary working directory and copy the
files there assuming that the source code was untarred in home directory.
mkdir ~/tutorial cd ~/tutorial cp ~/electrostatic-tools-0.1.0/examples/phbr.mol2 . cp ~/electrostatic-tools-0.1.0/examples/phbr.esp .
To perform a simple fit with default parameters invoke:
mult_fitter phbr.mol2 phbr.esp
In case everything went well no console output appears. This command prefrormed
the fit of atomic charges and multipoles of the phenylbromide molecule to the
reference MEP with default settings. Two files should appear in the working
directory: phbr.mmol and phbr.log. The first one contains the results - a
phenylbromide molecule with multipoles, the second one is a log with an additional
information describing the fit process. Your phbr.mmol file should look like the
following:
Molecule: /* Atom 1 Br */ Atom: 35 ( 0.9991, 0.0803, 0.1295 ) Multipoles: ( 0.9991, 0.0803, 0.1295 ) Monopole: -0.18347 Quadrupole: ( -0.81548, 0, 0, 0, -0.81548, 0, 0, 0, 1.631 ) /* Atom 2 C */ Atom: 6 ( 2.882, 0.1014, -0.0402 ) Multipoles: ( 2.882, 0.1014, -0.0402 ) Monopole: 0.27616 /* Atom 3 C */ Atom: 6 ( 3.5573, 1.3195, -0.1026 ) Multipoles: ( 3.5573, 1.3195, -0.1026 ) Monopole: -0.26482 /* Atom 4 C */ Atom: 6 ( 4.9466, 1.3334, -0.2343 ) Multipoles: ( 4.9466, 1.3334, -0.2343 ) Monopole: -0.11137 /* Atom 5 C */ Atom: 6 ( 5.6536, 0.132, -0.3014 ) Multipoles: ( 5.6536, 0.132, -0.3014 ) Monopole: -0.15636 /* Atom 6 C */ Atom: 6 ( 4.9736, -1.0845, -0.235 ) Multipoles: ( 4.9736, -1.0845, -0.235 ) Monopole: -0.11137 /* Atom 7 C */ Atom: 6 ( 3.5842, -1.1018, -0.1044 ) Multipoles: ( 3.5842, -1.1018, -0.1044 ) Monopole: -0.26482 /* Atom 8 H */ Atom: 1 ( 3.0135, 2.2588, -0.0508 ) Multipoles: ( 3.0135, 2.2588, -0.0508 ) Monopole: 0.18885 /* Atom 9 H */ Atom: 1 ( 5.478, 2.2803, -0.2855 ) Multipoles: ( 5.478, 2.2803, -0.2855 ) Monopole: 0.14652 /* Atom 10 H */ Atom: 1 ( 6.7358, 0.1442, -0.4058 ) Multipoles: ( 6.7358, 0.1442, -0.4058 ) Monopole: 0.14529 /* Atom 11 H */ Atom: 1 ( 5.5259, -2.0194, -0.2859 ) Multipoles: ( 5.5259, -2.0194, -0.2859 ) Monopole: 0.14652 /* Atom 12 H */ Atom: 1 ( 3.0612, -2.0529, -0.0547 ) Multipoles: ( 3.0612, -2.0529, -0.0547 ) Monopole: 0.18885 Bond: 1 - 2 : 1 Bond: 2 - 3 : a Bond: 3 - 4 : a Bond: 4 - 5 : a Bond: 5 - 6 : a Bond: 6 - 7 : a Bond: 2 - 7 : a Bond: 3 - 8 : 1 Bond: 4 - 9 : 1 Bond: 5 - 10 : 1 Bond: 6 - 11 : 1 Bond: 7 - 12 : 1 Orient-rules: rule: z "*" rule: a "[!#99]~[!#99]" rule: a "[!#99]#[!#99]" rule: b "[!#99]~[!#99]=,@[!#99]" rule: b "[!#99]=[!#99]~[!#99]" rule: c "[!#99](~[!#99])~[!#99]" rule: c "[!#99](=,@[!#99])~[!#99]" rule: d "[!#99]([!#99])([!#99])[!#99]" rule: b "[!#99](=[!#99])([!#99])[!#99]" rule: z "[!#99]([!#99])([!#99])([!#99])[!#99]" rule: a "[!#99](=[!#99])=[!#99]" rule: a "[!#99](=[!#99!#6])=[!#99]" rule: b "[!#99](~[!#99])(=[!#99])=[!#99]" rule: a "[#99]1[!#99]#[!#99]1" rule: e "[#99]1[!#99A]([!#99])=,@[!#99A]1" rule: e "[#99]1[!#99]-,=,@[!#99]1" /* Fitted 14 parameters with 6 constraints by 4617 points RMSD: 0.6277 kcal/mol */
It consists of three sections: the molecule description, the rules for orientation of the
multipoles and a comment.
We support C-style comments (/*comment */) in our custom file formats, so it is
possible to store any relevant information directly in the file with a molecule.
mult_fitter saves a fitted MEP description error and a number of parameters with
a number of applied constraints.
The molecule section consists of a description of atoms and bonds. The
bond format is intuitive. The atoms are stored as pairs of nucleus charges
with coordinates in Angstroms. Every atom may have a single multipole
expansion associated with it. The multipoles values are printed in atomic units.
The quadrupole matrix is printed in a row-by-row manner on a single line.
Note that the multipoles are shown in principal axes, so they can be easily
analysed.
The contents of the phbr.log file should be as the following:
Multipole fitter v 0.1.0 MEP Fitter initialized. Forcing tolopogical equivalency: 1 Tolopogical information will be recalculated: 0 Molecule with 12 atoms loaded. 20 atom and 0 group multipole positioning rules loaded. 4617 points of field loaded. 0 dummy atoms added. Created distance matrix 4617x14 Using SVD fitter. Created constraints matrix 6x14 Fitting Fit completed. Fitted 14 parameters with 6 constraints by 4617 points RMSD: 0.6277 kcal/mol ----------------- SVD analysis ----------------- Condition number cutoff: 1e+07 Condition number of the system: 99.1938 Singular values: 5.88127 2.25863 1.44742 1.21237 0.482768 0.195794 0.105907 0.0592907 1.26362e-15 2.02354e-16 6.8374e-17 1.13256e-17 5.90198e-18 4.81634e-20 Parameters: Z1 Qxx1 Qyy1 Z2 Z3 Z4 Z5 Z6 Z7 Z8 Z9 Z10 Z11 Z12 Right singular vectors (V**T): 0.516791 0.00489199 0.00489199 0.260871 ...
Along with the details of the fit this file contains a valuable information provided by
a SVD fitter: the condition number of a model matrix and a list of singular values
with right singular vectors. In this particular case the first shows that the charges
and the quadrupole were well-defined. The last two ones help analyse the system if
any problem with the fit stability occurs. The ”Parameters:” header and the ”V**T”
matrix are tab separated so they can be easily pasted in any spreadsheet processor
for further analysis.
If you try to rerun the calculation (note a different way to pass command line
arguments) it will terminate with the following error:
$mult_fitter -I phbr.mol2 -G phbr.esp -O phbr.mmol \ -L phbr.log Error: output molecule file exists. Aborting.
No Electrostatic Tools program will overwrite existing files to preserve any previous
results in case of a typo. To ignore this and overwrite the existing files pass a -f
key.
3.2 Multipole Placement
In the previuos example we used the default multipole placement policy. This policy
can be overriden by specifying a multipole placement rulefile. Let’s assume we
want to place a symmetric quadrupole on the bromine and a dipole on the
p-hydrogen atoms. Create a custom file with the rules and perform the fit
with these rules by invoking (note another way of passing the filenames):
cat > custom.rules << "EOF" Placement-rules: /* charge on any atom */ atom: "*" m /* charge + symmetric quarupole on bromine */ atom: "[Br]" mq* /* charge + dipole on p-hydrogen */ atom: "[#1]cccc[Br]" md EOF mult_fitter -Iphbr.mol2 -Gphbr.esp -p custom.rules \ -Ophbr.custom.mmol
phbr.custom.mmol contains the resulting molecule. Currently we support fitting of
dipoles with the fixed direction along a local Z-axis, so the dipole has two zero
components. The rulefile 1has a quite simple syntax. We specify a SMARTS pattern
of the desired atom (Note that hydrogens are matched by "[#1]" and NOT "[H]")
and a list of the required multipoles: m, d and q for a monopole, a dipole and a
quadrupole respectively with an asterisk (*) meaning a symmetry restriction on the
quadrupole. The patterns are matched in the same order as they go in the file. Each
subsequent rule overrides the previous ones, so the most general rules should go
before anything special. When no rulefile is specified the default, located in
__prefix__/share/electrostatic_tools/<version>/ placement.rules, is
used.
3.3 Topology Equivalence
For this tutorial we will need the meoh.* files from the examples directory.
By default mult_fitter preserves topology equivalence so the equivalent atoms get
the equivalent charges and multipoles fitted. This behaviour can be overriden by a -b
flag. For example we want to fit atomic charges for methanol molecule without
respect to its topology (namely the equivalence of hydrogen atoms within the -CH3
group).
mult_fitter -I meoh.mol2 -G meoh.esp -O meoh.b.mmol \ -p m.rules -b
Note that we are using our custom multipole placement rules without any multipoles
beyond monopole. The programs uses atomic charges as the source of topological
information. If two atoms of the same chemical element have equal charges, they are
considered equivalent. By inspecting the meoh.mol2 and meoh.b.mmol files one can
check that however the input file had a topologically symmetrical charges,
the output does not. On the contrary, if an input molecule has bad charges
assigned, the topologically symmetric Gasteiger charges may be recalculated
with the -r key to enforce the charge (and multipole) symmetry of the fit:
mult_fitter -I meoh.mulliken.mol2 -G meoh.esp \ -O meoh.r.mmol -p m.rules -r
3.4 Extra-Point Position Fit
The ep-fitter program is used to perform the optimization of the extra-point (EP)
positions. It places extra-points at atoms specified with SMARTS mask. We will
call these atoms EP-hosts. Lets perform the EP position optimizaion for a
m-fluorobrombenzene which is saved in mol36.mol2 and mol36.esp files in the
examples directory. We’ll place the extra-points on the bromine atom and
use a charge-only rulefile which can be found in the examples directory.
ep_fitter -I mol36.mol2 -G mol36.esp -p m.rules \ -M"[Br]"
The output was saved to the mol36.ep.mmol and mol36.ep.log files. The program
places the EP on the local Z-axis of the atom, defined by the orient rules, and then
searches for the position with the lowest MEP RMSD value. The optimization tooks
more time than a simple charge fit since it uses the Nelder-Mead simplex algorith for
the position optimization with the charge refit at every step. The program saves the
EP as the atom with a zero nuclear charge meaning a dummy, nonexistent,
atom.
The use of the Nelder-Mead algorithm looks strange when dealing with a single
parameter optimization, however we can optimize several EP centers simultaneously.
Lets try to add extra-points to the both bromine and fluorine, This time
we will need to increase a maximum iteration limit for the optimization
to converge. We have also switched to a faster charge fitting algorithm to
save some time. Additionally we specified a different file for the output.
ep_fitter -I mol36.mol2 -G mol36.esp -p m.rules \ -M"[Br]" -M"[F]" -a FullPivLU -m 150 \ -O mol36.2ep.mmol
If you investigate the result you will notice, that the EP-Br distance converged to 1.3
Å, while the EP-F distance became -5 Å. The negative value of the distance means
that EP has penetrated its host atom and traversed inside the molecule. In this case,
-5 Åactually means that it traversed the whole molecule and stopped at the reference
grid border. This behaviour is normal for the fluorines since they do not need any
special anisotropy treatment.
One more thing should be mentioned about the EP position optimization
possibilities. Since we’re using the same code for the charge fitting, it is possible to fit
simultaneously both multipoles and EP positions (with relevant placement rules set).
It is even possible to place multipoles on the extra-point centers and optimize their
positions, however note, that internally EPs are stored as Einsteinium ("[#99]",
because matching "[#0]" doesnot work). When the multipole placement rules tell
the fitter to add the multipoles on the EP-host atom it ignores this rule, leaving the
host with charge only, however this behaviuor can be overriden by the -k
switch.
3.5 Converting MMol to Common Format
After we fitted the charges or multipoles, we can convert the resulting .mmol file into
any common chemical file format supported by OpenBabel with the mmol2mol
program. Try this:
By default it strips all multipoles and saves the molecule as a TRIPOS MOL2 file, but
this behaviour can be overriden with the -O and -t flags to change the output file
and its format if automatic extention based guessing fails. You can also try MCC
conversion options to preserve multipole data, however this part is still under
research and is subject to change.
3.6 Using Script Bindings
To analyse fit results you can also use script language bindings. Unfortunately
currently we do not provide any reference documentation, so you’ll have to look
into source code for reference. We’ll provide a simple example python script
here. This script uses the phbr.mmol file generated earlier in this tutorial.
import openbabel import ElectrostaticTools # read molecule with multipole support from file mol = ElectrostaticTools.GeneralMultipoledMolecule() mol.readMe("phbr.mmol") # save it as MOL2 # note: GeneralMultipoledMolecule is OBMol subclass # so we can use any OpenBabel API conv = openbabel.OBConversion() conv.SetOutFormat("mol2") conv.WriteFile(mol, "test.mol2") # we can iterate other the atoms for atom in openbabel.OBMolAtomIter(mol): print (atom.GetAtomicNum()) # print molecule in MMOL format print (mol.toString()) # check if multipole expansion exists on the first # atom and print the monopole and quadrupole value if (mol.hasMultipoles(1)) : mult = mol.GetRawMultipoles(1) if (mult.hasMonopole()) : print ("\nMonopole value: ", mult.monopole().value()) if (mult.hasQuadrupole()) : print ("\nMonopole value: ", mult.quadrupole().value().toString()) # or simply print the expansion as is print ("\nMultipoles on the first atom:") print (mult.toString()) # or we can get oriented multipoles print ("\nOriented multipole on the first atom:") print (mol.GetMultipoles(1).toString()) # or we can get raw multipoles from atom mult = ElectrostaticTools.Multipoles( mol.GetAtom(2).GetData("Multipoles") ) print ("\nMultipoles from atom:") print (mult.toString())
4 Program Overview
4.1 General notes
All programs included in the package are non-interactive command line tools, written
with UNIX philosophy in mind – they are silent if everything is going fine, however
some noncritical messages can be logged. All tasks are formulated with command line
arguments. Thanks to the boost::program_options library there are several ways to
pass an argument to the program. The following invocations are identical:
progname --long-option arg progname --long-option=arg progname -l arg progname -larg
However some constructions are NOT available, such as:
progname --no-long-option progname --with-long-option progname --disable-long-option # etc ... progname -l=arg
Some arguments are considered essential for the program execution and can be
specified without a key. In this case the order of such keyless options is important.
For example:
mult_fitter molecule.mol2 grid.esp # is fine mult_fitter molecule.mol2 grid.esp -f # is fine mult_fitter -c 1e9 molecule.mol2 grid.esp # is fine mult_fitter grid.esp molecule.mol2 # will terminate with an error
4.2 mult_fitter
The mult_fitter program performs fitting of the atom-centered multipoles to the
specified reference molecular electrostatic potential (MEP). The MEP is specified in
a form of AMBER .esp file. The input molecule can be specified in any chemical
format understood by the OpenBabel library. The result is saved into our plan-text
.mmol file format. Multipole positioning is controlled by SMARTS patterns which are
read from a separate file. The multipoles are fixed in the orientation regarding the
neighboring atoms which is controlled through another file. It is possible to constrain
the topological equivalence of atoms (which can be precisely controlled) and
the symmetry of quadrupoles. Addition of dummy multipole centers in the
geometrical center of atom groups specified by a SMARTS pattern is also
possible.
Some of possible invocation patterns:
mult_fitter <input> <MEP .esp> [output] [options] mult_fitter -I <input> -G <MEP .esp> -O <output> \ [options]
Generic options:
-
--version
- print the program name and version and exit.
-
--help
- print a help message to console and exit.
Input control:
-
-I, --input < arg > required
- an input molecule file. This parameter is also
read as the first argument without a key, so the key can be omitted.
-
-t, --filetype < arg >
- an input molecule file type specified as common
extenson for this filetype. Generally the filetype is guessed by the file
extension (GUESS option). This key overrides this guess.
-
-G, --grid < arg > required
- a reference MEP in the .esp format. This
parameter is also read as the second argument without a key, so the key
can be omitted. Atomic coordinates in the .esp files are ignored, but the
total charge is used as a constraint in the fitting procedure.
-
-p, --placement-rules < arg >
- multipole placement rules specified in
special format. The multipoles are placed according to SMARTS patterns.
Note that OpenBabel SMARTS are different from Daylight SMARTS
(see http://openbabel.org/wiki/SMARTS). See the format description for
more info on how to setup custom rules.
-
-o, --orient-rules < arg >
- multipole
orient rules. The multipoles are placed according to SMARTS patterns.
Note that OpenBabel SMARTS are different from Daylight SMARTS
(see http://openbabel.org/wiki/SMARTS). See the format description for
more info on how to setup custom rules.
Output control:
-
-O, --output < arg >
- an output molecule file. This parameter is also read
as the third argument without a key, so the key can be omitted. The
default name is generated by replacing the last file extension from the
input file name with mmol. In order to save previous results, the program
will terminate if the output file exists. The molecule is saved in a custom
plain-text format which was designed to support atomic multipoles. See
the format description for more info.
-
-L, --log < arg >
- a log file. The default name is generated by replacing the
last file extension from the input file name with the log. In order to save
previous results, the program will terminate if the log file exists. The
log contains important information about program workflow and some fit
data. It is saved as a plain-text.
-
-f, --force-output
- a switch to overwrite the output and log files if they are
already present.
MEP fit control:
-
-a, --algorithm < arg >
- a MEP fit algorithm selector. Valid values
are: SVD, LLT, LDLT, PartialPivLU, FullPivLU, HouseholderQR,
ColPivHouseholderQR, FullPivHouseholderQR. The SVD algorithm uses
the method published by Sigfridson and Ryde (J. Comput. Chem. 1998,
19(4), 377). It works with a model matrix without raising it to the power
of two, thus increasing the stability of the fit. The other methods deal
with the squared model matrix and a Lagrange constraints. The details
about them can be found in Eigen3 documentation.
-
-c, --cutoff < arg >
- a condition value cutoff (the largest singular value
divided by the smallest one) used in the SVD pseudoinversion procedures
to eliminate statistical noise. This parameter is used only when fitting
with the SVD fitter. The default value is 107.
-
-r, --recalculate-topology
- a flag to force fitter to recalculate atomic
charges used to force the equivalency of the topologically equivalent atoms.
The atoms are forced to have equal charges and multipoles if they belong
to the same chemical element and have the equal partial charges assigned.
If this flag is set, the Gasteiger charges are calculated and used for this
purpose since they are topologically symmetrical. Note that sometimes
there are not enouth digits in a charge field of the input molecule file
and non-equivalent charges have the same charge, for example meta- and
para- hydrogens and sometimes carbons in monosubstituted benzenes in
the MOL2 file format. Generally turning on this flag is a good idea if you
are not controlling your input charges manually.
-
-b, --break-equivalency
- a flag to disable the equivalency constraints. The
only constraints added with this flag is total charge and quadrupole
symmetry constraints.
4.3 ep_fitter
The ep_fitter program fits the extra-point (EP) charge positions and
atom-centered multipoles with the specified reference molecular electrostatic
potential (MEP). The MEP is specified in a form of AMBER .esp file. The input
molecule can be specified in any chemical format understood by the OpenBabel
library. The result is saved into our plan-text .mmol file format. Multipole
positioning is controlled by SMARTS patterns which are read from a separate
file. The multipoles are fixed in the orientation regarding the neighboring
atoms which is controlled through another file. It is possible to constrain the
topological equivalence of atoms (which can be precisely controlled) and
the symmetry of quadrupoles. Addition of dummy multipole centers in the
geometrical center of atom groups specified by a SMARTS pattern is also
possible. The extra-points are added to the host atoms specified by SMARTS
patterns.
This program uses the same code as the mult_fitter for multipole and charge
fitting so most of the options are exactly the same, however all options are provided
here for consistency. The nonlocal EP position optimization runs on top of the linear
charge and multipole fitting. The Nelder-Mead simplex search local optimization
algorithm is used for this purpose.
Some of the possible invocation patterns:
ep_fitter <input> <MEP .esp> -M"[Cl,Br,I]" \ [output] [options] ep_fitter -I <input> -G <MEP .esp> -O <output> \ -M"[Cl,Br,I]" [options]
Generic options:
-
--version
- print the program name and version and exit.
-
--help
- print a help message to console and exit.
Input control:
-
-I, --input < arg > required
- an input molecule file. This parameter is also
read as the first argument without a key, so the key can be omitted.
-
-t, --filetype < arg >
- an input molecule file type specified as common
extenson for this filetype. Generally the filetype is guessed by the file
extension (GUESS option). This key overrides this guess.
-
-G, --grid < arg > required
- a reference MEP in the .esp format. This
parameter is also read as the second argument without a key, so the key
can be omitted. Atomic coordinates in the .esp files are ignored, but the
total charge is used as a constraint in the fitting procedure.
-
-p, --placement-rules < arg >
- multipole placement rules specified in
special format. The multipoles are placed according to SMARTS patterns.
Note that OpenBabel SMARTS are different from Daylight SMARTS
(see http://openbabel.org/wiki/SMARTS). See the format description for
more info on how to setup custom rules.
-
-o, --orient-rules < arg >
- multipole
orient rules. The multipoles are placed according to SMARTS patterns.
Note that OpenBabel SMARTS are different from Daylight SMARTS
(see http://openbabel.org/wiki/SMARTS). See the format description for
more info on how to setup custom rules.
Output control:
-
-O, --output < arg >
- an output molecule file. This parameter is also read
as the third argument without a key, so the key can be omitted. The
default name is generated by replacing the last file extension from the
input file name with mmol. In order to save previous results, the program
will terminate if the output file exists. The molecule is saved in a custom
plain-text format which was designed to support atomic multipoles. See
the format description for more info.
-
-L, --log < arg >
- a log file. The default name is generated by replacing the
last file extension from the input file name with the log. In order to save
previous results, the program will terminate if the log file exists. The
log contains important information about program workflow and some fit
data. It is saved as a plain-text.
-
-f, --force-output
- a switch to overwrite the output and log files if they are
already present.
MEP fit control:
-
-a, --algorithm < arg >
- a MEP fit algorithm selector. Valid values
are: SVD, LLT, LDLT, PartialPivLU, FullPivLU, HouseholderQR,
ColPivHouseholderQR, FullPivHouseholderQR. The SVD algorithm uses
the method published by Sigfridson and Ryde (J. Comput. Chem. 1998,
19(4), 377). It works with a model matrix without raising it to the power
of two, thus increasing the stability of the fit. The other methods deal
with the squared model matrix and a Lagrange constraints. The details
about them can be found in Eigen3 documentation.
-
-c, --cutoff < arg >
- a condition value cutoff (the largest singular value
divided by the smallest one) used in the SVD pseudoinversion procedures
to eliminate statistical noise. This parameter is used only when fitting
with the SVD fitter. The default value is 107.
-
-r, --recalculate-topology
- a flag to force fitter to recalculate atomic
charges used to force the equivalency of the topologically equivalent atoms.
The atoms are forced to have equal charges and multipoles if they belong
to the same chemical element and have the equal partial charges assigned.
If this flag is set, the Gasteiger charges are calculated and used for this
purpose since they are topologically symmetrical. Note that sometimes
there are not enouth digits in a charge field of the input molecule file
and non-equivalent charges have the same charge, for example meta- and
para- hydrogens and sometimes carbons in monosubstituted benzenes in
the MOL2 file format. Generally turning on this flag is a good idea if you
are not controlling your input charges manually.
-
-b, --break-equivalency
- a flag to disable the equivalency constraints. The
only constraints added with this flag is total charge and quadrupole
symmetry constraints.
EP position fit control:
-
-M, --host-mask < arg >
- the SMARTS mask of the EP host atoms. The
key is repeatable so multiple masks can be specified. The EP is placed on
the local Z-axis specified by the orientation rules and it is ensured that the
EP is located ”outside” of the molecule. By default the multipoles, placed
on the EP hosts are removed, since it’s strange to have two anisotropic
enhancements for a single atom.
-
-x, --fixed-position < arg >
- fixed EP distance from halogen atom in
Angstroms. No optimization will be applied. Overrides any optimization
option.
-
-d, --init-position < arg >
- the initial distance between EP and its host
atom in Angstroms for optimization procedure. The default value is 1.5
Å.
-
-s, --init-step < arg >
- the initial step of EP-host distance in Angstroms
for the optimization procedure. Positive values mean the increase of the
distance while the negative ones mean the decrease. The default value is
0.1 Å.
-
-e, --precision < arg >
- a required position precision in Angstroms. Short
name is abbreviated from the word ”epsilon”. Technically, this is a
maximum simplex radius. The default value is 10-3 Å.
-
-m, --max-steps < arg >
- the maximum number of optimization steps. The
search is stopped at this point and the failure is reported in log. The
default value is 100.
-
-k, --keep-multipoles
- a flag to override removal of multipoles from EP
hosts.
4.4 mmol2mol
The mmol2mol program converts .mmol files t common chemical formats with respect
to the atomic multipoles in these files.
Some of possible invocation patterns:
mmol2mol <options> mmol2mol <in_mol> <out_mol> [options]
Generic options:
-
--version
- print the program name and version and exit.
-
--help
- print help message to console and exit.
Input control:
-
-I, --input < arg > required
- an input molecule file in the .mmol format.
This parameter is also read as the first argument without a key, so the
key can be omitted.
Output control:
-
-O, --output < arg >
- an output molecule file. This parameter is also read
as the second argument without a key, so the key can be omitted. The
default name is generated by replacing the last file extension from the input
file name with ”mol2” and the molecule is saved in a TRIPOS MOL2 file
format. In order to save the previous results, the program will terminate
if the output file exists.
-
-t, --filetype < arg >
- an output molecule filetype, specified as a common
extenson for this filetype. Generally the filetype is guessed by the output
file extension (”GUESS” option). This key overrides this guess.
-
-f, --force-output
- a switch to overwrite the output files if it is already
present.
Conversion control:
NOTE: The further options work, however the work on MCC is still in progress and
not published, so the behaviour may change in future versions. Use it on your own
risk.
-
-M, --mcc-mask < arg >
- the multipoles on the atoms matching this mask
will be substituted by a multipole charge cluster (MCC). This cluster is
composed of a set of several extra-point charges (2-6) and in combination
with the charge of the host atom creates a MEP distribution, analogous
to the multipolar one. The key is repeatable so multiple masks can be
specified.
-
-r, --radius < arg >
- the MCC radius in Angstroms as the distance between
the host atom and the extra-points. The default is 0.1 Å.
-
-d, --ignore-dipole
- do not convert atomic dipoles to the MCC.
-
-d, --ignore-quadrupole
- do not convert atomic quadrupoles to the MCC.
5 File Formats
5.1 General Notes
All the following file formats support C-style comments (/*comment */) so any
additional information can be stored next to the data in an arbitrary format. The
comment parsers are not very smart so do not nest your comments. The spaces,
newlines and tabulation characters are ignored, so a fancy text alighning can be
achieved. We prefer to save SMARTS patterns in quotes. These quotes are required
by the format, so we can check that a pattern was specified and we’re not reading
something different.
The files are separated in sections. Every section starts with a header ending with a
colon sign and ends with the beginning of the next section.
5.2 Multipole Orient Rules
Multipole orient rules controls the orientation of a local coordinate frame for each
atom. The rules start with a common ”Orient-rules:” header and contain
records of individual rules. The records are passed from top to bottom, with
the latter overriding the former, so the ordering is important. The first one
should be something general with very specific ones at the bottom of the list.
Orient-rules: rule: z "*" rule: a "[!#99]~[!#99]" rule: b "[!#99]=[!#99]~[!#99]" rule: c "[!#99](~[!#99])~[!#99]" rule: d "[!#99]([!#99])([!#99])[!#99]" rule: z "[!#99]([!#99])([!#99])([!#99])[!#99]" rule: e "[#99]1[!#99]-,=,@[!#99]1"
Each rule starts with the ”rule:” keyword, followed by a letter, followed by a
SMARTS pattern. The quotes around the SMARTS pattern are mandatory. The
order of atoms in SMARTS is important in most cases. The first atom is a center of
the multipole expansion and the local frame orign, the meaning of the others are
determined by the letter, which encodes the rule type. Note that we use
"[#99]" internally as a dummy atom, because SMARTS like "[#0]" or
"[#200]" do not work. That’s why the orientation rule’s SMARTS patterns
look a little strange. Also note, that if your molecule contains Einsteimium
(which is hardly the case), you should temporarly change it to something
different.
-
- z Only the first atom is important. This rule means that we do not care
about the local frame orientation. For example it is hard to pick sensible
orientation for tetrary carbons. Identity matrix is used as the coordinate
transformation matrix.
-
- a The first two atoms are important. This is the rule for linear nonconjugated
fragments such as monovalent atoms or alkynes. The Z-axis is directed
from the second atom to the first. The X- and Y- axes are undefined and
picked through a vector product of the local Z-axis and the global axes.
-
- b The first three atoms are important. This is the rule for conjugated linear
fragments, or the fragments, where we can suspect any type of interaction
with the nearest neighbour, or for the atoms with double bonds. In this
case the Z-axis is directed from the second atom to the first. The X-axis is
perpendicular to the plane, defined by the first three atoms in SMARTS
and the Y-axis is perpendicular to the local X- and Z- axes.
-
- c The first three atoms are important. This rule is for bivalent linkers like
ether group. The Z-axis directed along the bisector of 2-1-3 angle and
poits from the sharp end of this angle ”outside” of the molecule. The
X-axis is perpendicular to the plane, defined by the first three atoms of
the SMARTS and the Y-axis is perpendicular to the X- and Z- axes.
-
- d The first four atoms are important. This rule is for trivalent atoms like amine
nitrogen.
- In the case of a pyramidal configuration of the first atom, the Z-axis
points out of the top of the pyramid in the direction, formed as sum
of normalized bond vectors, pointing to the top of the pyramid. The
X-axis is defined as a vector product of the Z-axis and the 2→1 bond
vector. The Y-axis is a vector product of the X- and Z- axes.
- In the case of a planar configuration of the first atom, the X-axis is
the 2→1 bond vector, the Z-axis is a vector product of the X-axis
and the 3→1 bond vector, so it points out of the plane. The Y-axis
is perpendicular to the both X- and Z- axes.
-
- e The first three atoms are important. This is special rule for the dummies. The
X-axis is defined as the vector from the second atom to the first one. The Z-axis
is perpendicular to the plane defined by the first three atoms. The Y-axis is
perpendicular to the both X- and Z- axes.
5.3 Multipole Placement Rules
The Multipole placement rules format is easy to read and modify. The rules start
with a common ”Placement-rules:” header. There are two types of records: ”atom”
and ”group”. The records are passed from the top to the bottom, with the latter
overriding the former, so the ordering is important. The first one should be
something general with very specific ones at the bottom of the list. See the example
with an ”any atom”, followed by a ”halogen”, followed by the ”halogen, connected to
an aromatic moiety”.
Placement-rules: atom: "*" m atom: "[Cl,Br,I]" mdq* atom: "[Cl,Br,I]a" mq* group: "c1ccccc1" mdq*
Each atom rule starts with a ”atom:” keyword, followed by a SMARTS pattern in
quotes, followed by multipole flags. The quotes around the SMARTS pattern are
mandatory. The multipole flags can be any combination of ”m”, ”d”, ”q” and ”*”,
meaning a monopole, a dipole, a quadrupole and a symmetry restriction of the
quadrupole respectively. When a SMARTS match of an atom rule happens,
Electrostatic Tools programs will add the specified multipoles to the first atom of the
SMARTS pattern.
Group rules start with ”group:” keyword, followed by a SMARTS pattern in
quotes, followed by the multipole flags. The formatting and properties are
analogous the to atom rules. When a group SMARTS match happens, a
program will add a dummy atom center to the geometrical center of all
SMARTS atoms and place the specified multipoles on this dummy center. The
dummy center becomes connected with the first two atoms in the group’s
SMARTS.
5.4 Multipole Molecule
The ”mmol” format was designed to store molecules with associated multipoles. It
consists of the two sections: the molecule with atoms and bonds, and the
multipole orient rules part. The latter is described in the corresponding
section above. The molecule section contains ”Atom” and ”Bond” records.
Molecule: Atom: 6 ( 1.1057, 0.0178, -0.0171 ) Multipoles: ( 1.1057, 0.0178, -0.0171 ) Monopole: -0.136 Atom: 8 ( 2.5213, 0.0064, -0.0264 ) Multipoles: ( 2.5213, 0.0064, -0.0264 ) Monopole: -0.26592 Dipole: ( 0, 0, -0.4019 ) Quadrupole: ( -0.82922, 0, 0, 0, 1.0774, 0, 0, 0, -0.24822 ) Atom: 1 ( 0.7455, 0.9809, -0.3871 ) Multipoles: ( 0.7455, 0.9809, -0.3871 ) Monopole: 0.062576 Atom: 1 ( 0.7455, -0.1514, 1.0007 ) Multipoles: ( 0.7455, -0.1514, 1.0007 ) Monopole: 0.062576 Atom: 1 ( 0.7398, -0.7799, -0.6679 ) Multipoles: ( 0.7398, -0.7799, -0.6679 ) Monopole: 0.062576 Atom: 1 ( 2.8166, 0.7242, 0.5592 ) Multipoles: ( 2.8166, 0.7242, 0.5592 ) Monopole: 0.2142 Bond: 5 - 1 : 1 Bond: 3 - 1 : 1 Bond: 2 - 1 : 1 Bond: 2 - 6 : 1 Bond: 1 - 4 : 1 Orient-rules: rule: z "*" rule: a "[!#99]~[!#99]" rule: c "[!#99](~[!#99])~[!#99]"
The atom record starts with the ”Atom:” keyword followed by a nuclear charge (a.
u.) and nuclear coordinates in brackets, separated by a comma (in Angstroms).
Optionally it can contain a ”Multipoles” field.
The ”Multipoles” record starts with the ”Multipoles:” keyword followed by
the coordinates of the expansion center in brackets, separated by a comma
(in Angstroms). Next, it contains three optional fields: a ”Monopole:”, a
”Dipole:” and a ”Quadrupole:” records, followed by the corresponding
multipole moment value (in a. u.). Tensor values are written in brackets
with components separated by a comma. A single ”Multipoles” record has a
single internal coordinate system. The multipoles are written in terms of the
principal axes of the quadrupole. The orientation of the local coordinate
frame is governed by the orient rules, recorded in its own section of file. We
use the following formulae to calculate the electrostatic potential from the
multipoles:
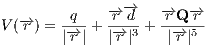
, where 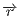 corresponds to radius-vector from the center of the multipole expansion
to a potential estimation point,
corresponds to radius-vector from the center of the multipole expansion
to a potential estimation point, 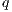 ,
, 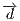 and
and 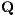 correspond to the charge, the
dipole moment vector and the quadrupole moment matrix, transformed to the global
coodinate frame.
correspond to the charge, the
dipole moment vector and the quadrupole moment matrix, transformed to the global
coodinate frame.
The bond record starts with the ”Bond:” keyword, followed by the first atom index,
followed by a ”minus” sign, followed by the second atom index, followed by a colon
sign, followed by a bond order value: (”1”, ”2”, ”3” or ”a” for single, double, triple
aromatic bonds respectively).