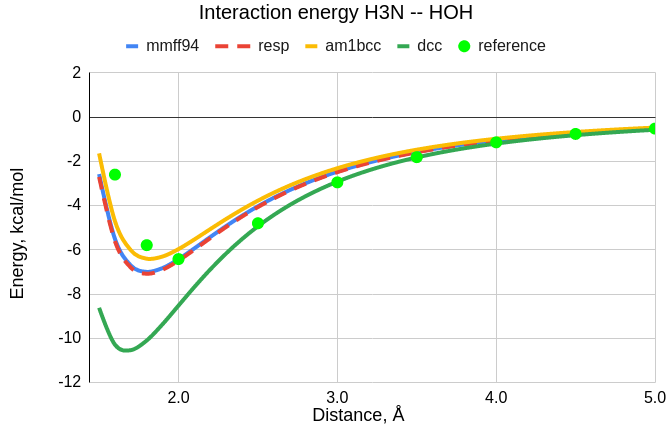
Use of DCC (Extra-point) model within GAFF force field.
In this section we briefly review application of MEP-fitted Distributed Charge Cluster model
(DCC, see sections EP-placement and EP-fit of
Tutorial for more info) for molecular simulations using
mdgx molecular dynamics system.
We calculate free energy of a bimolecular complex, using different charge schemes
for electrostatic modelling within GAFF force field, and compare results.
RESP, AM1-BCC, MMFF94 and DCC charge models, aimend to reproduce RHF/6-31G*,
are compared to the reference ab initio data,
calculated at MP2/aug-cc-pVTZ level with frozen monomer geometries.
Additional requirements to reproduce the following guide are:
OpenBabel and AmberTools20 software packages.
Specifically, obabel, respgen, resp,
antechamber, tleap, parmchk2 and mdgx
external utilities are used in this tutorial.
You can reproduce the results obtained with the set of scripts and files supplied with the source code.
For the reference input and output files are provided result.tar.xz .
As a model system, complex of ammonia with hydrogen is used (Figure 1). In this complex ammonia's nitrogen and water's hydrogen forms explicit hydrogen bond which is the case for illustrative demonstration of difference in energy profiles, obtained using different charge models.
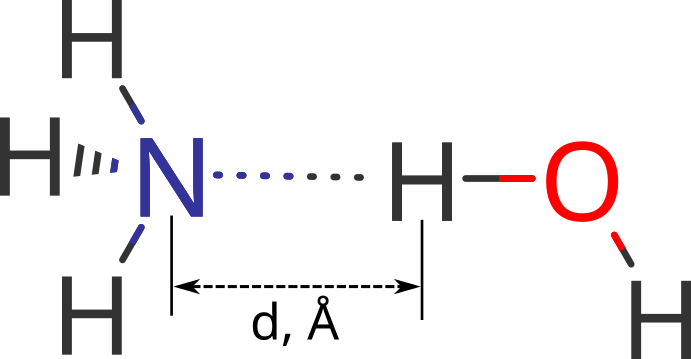
Figure 1. Relative orientation of ammonia and water in a complex; d=1.6, 1.8, 2.0, 2.5, 3.0, 3.5, 4.0, 4.5, 5.0 Å
Distance between Nitrogen and Hydrogen variates from 1.6 to 5 Angstroms, including both values, with a step of 0.2 Angstroms. Overall, 9 molecular systems are examined.
Original molecules files may be found in examples/Appendix_A directory.
Those files contain ammonia molecule (H3N.mol2) and
a set of files with pre-oriented and pre-positioned water molecules
(H2O_%05.2f.mol2). Esp file for ammonia calculated
in 6-31G* basis is included (H3N.esp).
This directory also contains ready2use shell scripts implying
utilities listed above are in your $PATH.
1. Charges calculation
At the first stage, we calculate charges for ammonia molecule
which are required to be assigned to the corresponding molecule
before generating .prmtop and .prmcrd files with force field
parameters. As it was mentioned before, all listed scripts may
be found in examples/Appendix_A directory.
Listing 1: MMFF94 charges calculation procedure (calc_mmff94_charges.sh)
# calculate mmff94 charges
obabel \
"original/mol2/H3N.mol2" -i mol2 \
-O "result/H3N_mmff94.mol2" -o mol2 \
--partialcharge mmff94
Listing 2: RESP charges calculation procedure (calc_resp_charges.sh)
# calculate RESP charges
# prepare input files for 2-step resp calculation
antechamber \
-pf y \
-i "original/mol2/H3N.mol2" -fi mol2 \
-o "tmp/H3N.ac" -fo ac \
-dr n -j 0
respgen -i "tmp/H3N.ac" -o "tmp/H3N.resp1.in" -f resp1
respgen -i "tmp/H3N.ac" -o "tmp/H3N.resp2.in" -f resp2
# perform 2-step resp calculation
resp \
-O \
-i "tmp/H3N.resp1.in" \
-o "tmp/H3N.resp1.out" \
-t "tmp/H3N.resp1.q" \
-s "tmp/H3N.resp1.esout" \
-p "tmp/H3N.resp1.punch" \
-e "original/esp/H3N.esp"
resp \
-O \
-i "tmp/H3N.resp2.in" \
-o "tmp/H3N.resp2.out" \
-t "tmp/H3N.resp2.q" \
-q "tmp/H3N.resp1.q" \
-s "tmp/H3N.resp2.esout" \
-p "tmp/H3N.resp1.punch" \
-e "original/esp/H3N.esp"
# assign computed charges
antechamber \
-pf y \
-i "original/mol2/H3N.mol2" -fi mol2 \
-o "result/H3N_resp.mol2" -fo mol2 \
-c rc -cf "tmp/H3N.resp2.q"
Listing 3: AM1-BCC charges calculation procedure (calc_am1bcc_charges.sh)
# calculate AM1-BCC charges
antechamber \
-pf y \
-nc 0 \
-i "original/mol2/H3N.mol2" \
-fi mol2 \
-o "result/H3N_am1bcc.mol2" \
-fo mol2 \
-j 4 \
-c bcc \
-dr n
rm -f sqm.in sqm.out sqm.pdb
Listing 4: DCC charges calculation procedure (calc_dcc_charges.sh)
# calculate DCC charges in two steps
# first, add External Points on sigma bonds
# of original ammonia molecule
# so it would be like H-ep1-N-ep2
# for every H-N bond
mmol2mol \
-f \
--input "original/mol2/H3N.mol2" \
--output "tmp/H3N+ep.mol2" \
--ep-place-on-bond "1 1 2 0.3" \
--ep-place-on-bond "1 1 2 -0.3" \
--ep-place-on-bond "1 1 3 0.3" \
--ep-place-on-bond "1 1 3 -0.3" \
--ep-place-on-bond "1 1 4 0.3" \
--ep-place-on-bond "1 1 4 -0.3"
# second, fit the system to the reference ESP
# manually setting topology equality constraints
# for DCC points
ep_fitter \
-f \
--input "tmp/H3N+ep.mol2" \
--output "tmp/H3N_dcc+ep.mmol" \
--grid "original/esp/H3N.esp" \
--placement-rules "original/rules/m.rules" \
--refit \
--break-equivalency \
--constraint "0 1 -1 0 0 0 0 0 0 0" \
--constraint "0 0 1 -1 0 0 0 0 0 0" \
--constraint "0 0 0 0 1 0 -1 0 0 0" \
--constraint "0 0 0 0 1 0 0 0 -1 0" \
--constraint "0 0 0 0 0 1 0 -1 0 0" \
--constraint "0 0 0 0 0 1 0 0 0 -1"
# convert .mmol to .mol2
mmol2mol \
-f \
--input "tmp/H3N_dcc+ep.mmol" \
--output "result/H3N_dcc+ep.mol2"
DCC charges extraction
The H3N_dcc.mol2 file need further processing
to become suitable for AmberTools utilities. Since Extra
Points are dummy atoms and have no real atomic number,
they can not be processed by antechamber,
which is used for GAFF atom types assignment. Thus, EPs
must be removed while atomic charges should be kept the same.
It can be done either manually by deleting lines containing
Xx from H3N_dcc.mol2 and changing
atoms number in the file header or using the following script.
In result, one should obtain the set of files (located in
result directory), containing ammonia molecule
with charges assigned via different schemes:
-
H3N_mmff94.mol2 -
H3N_resp.mol2 -
H3N_am1bcc.mol2 -
H3N_dcc.mol2
Water molecule preparation
Water molecule(s) were prepared using the folowing procedure. For the sake of simplicity, TIP3P water model was used.
Listing 6: Water molecules preparation procedure (prepare_water.sh)
# assign TIP3P params to each water molecule
for d in 01.6 01.8 02.0 02.5 03.0 03.5 04.0 04.5 05.0; do
water_in_prefix="original/pdb/H2O_term_d_${d}"
water_out_prefix="result/H2O_term_d_${d}"
tleap_in="tmp/H2O_term_d_${d}.tleap.in"
cp "prepare_water_template.tleap.in" "${tleap_in}"
sed -i "s,H2O_in,${water_in_prefix},g" "${tleap_in}"
sed -i "s,H2O_out,${water_out_prefix},g" "${tleap_in}"
tleap -s -f "${tleap_in}"
done
Force field parametes asignment
At the next stage, we assign forcefield (GAFF) parameters to our molecules.
To do that, first we need to assign GAFF atomic types to ammonia molecule.
Atomic types assignment for small molecules may be performed via
antechamber utility.
Listing 7: GAFF types assignment procedure (assign_gaff_types.sh)
# for all files with charges calculated
for i in H3N_mmff94 H3N_resp H3N_am1bcc H3N_dcc; do
# generally, this first step is unnecessary
# but performed to ensure that partial charges
# are marked as user-defined
# for that we extract calculated atomic charges
# to separate file(s) and then pass them
# on the next step as manually-defined
antechamber \
-pf y \
-i "result/${i}.mol2" -fi mol2 \
-o "tmp/${i}.mol2" -fo mol2 \
-c wc -cf "tmp/${i}.mol2.q" \
-dr n -j 0
# at the second step assign GAFF types and set
# previously extracted atomic charges
antechamber \
-pf y \
-i "result/${i}.mol2" -fi mol2 \
-o "result/${i}.gaff.mol2" -fo mol2 \
-c rc -cf "tmp/${i}.mol2.q" \
-at gaff \
-dr y -j 4
done
Next, we explicitly obtain all force field parameters values using
parmchk2 utility for all kinds of ammonia and water molecules.
Listing 8: GAFF parameters assignment procedure (get_gaff_parameters.sh)
# use parmchk2 to get GAFF parameters
# for every ammonia molecule
for i in H3N_mmff94 H3N_resp H3N_am1bcc H3N_dcc; do
echo "${i}"
parmchk2 \
-i "result/${i}.gaff.mol2" -f mol2 \
-o "result/${i}.gaff.frcmod" \
-s 1 \
-a Y \
-w Y
done
# same for every water molecule
for i in 01.6 01.8 02.0 02.5 03.0 03.5 04.0 04.5 05.0; do
echo "${i}"
parmchk2 \
-i "result/H2O_term_d_${i}.gaff.mol2" -f mol2 \
-o "result/H2O_term_d_${i}.gaff.frcmod" \
-s 1 \
-a Y \
-w Y
done
Preparation of mdgx input files
Combining ammonia and water molecules into complex
Now we can use molecules with assigned types an obtained force
field parameters to prepare ammonia+water complex via
tleap utility. H2O, H3N and COMPLEX entries should
be replaces with a corresponding input water molecule filename,
input ammonia molecule filename and output complex filename
(for details see prepare_complex.sh). As a result,
we obtain 3 files: ammonia+water complex in .mol2 format,
.prmtop and .prmcrd files to use with mdgx.
Listing 9: Tleap input file example for combining ammonia and war (prepare_complex_template.tleap.in)
source leaprc.gaff
source leaprc.water.tip3p
loadamberparams H2O.gaff.frcmod
loadamberparams H3N.gaff.frcmod
h2o = loadmol2 H2O.gaff.mol2
check h2o
h3n = loadmol2 H3N.gaff.mol2
check h3n
c = combine { h2o h3n }
check c
saveamberparm c COMPLEX.gaff.prmtop COMPLEX.gaff.prmcrd
savemol2 c COMPLEX.gaff.mol2 0
quit
Preparation of EP description suitable for mdgx
mdgx uses its own format to describe External Points
postions; EPs are only used through simulaton process -
input and output molecule files do not contain them in any form.
To set up EPs positions, one generally must specify uneque EP name, type of rule to position EP, distance between EP and a pivot atom and partial charge EP bears. It should be noted that EPs considered having zero mass and VdW radius.
Part of file used in this work (dcc_template.mdgx.rules) is listed below.
Listing 10: Tleap input file example for combining ammonia and war (dcc_template.mdgx.rules)
&rule
epnameE1,
style 1,
frame1N,
frame2H,
v12 DIST1,
q Q1,
sig 0.0,
eps 0.0,
residueLIG,
&end
&rule
epnameE2,
style 1,
frame1N,
frame2H,
v12 DIST2,
q Q2,
sig 0.0,
eps 0.0,
residueLIG,
&end,
-
epnamedefines EP name -
styledefines a certain rule to position EP; 1 means placement along the bond between two atoms (specified byframerules) -
v12defines distance (in this case, relative toframe1atom) -
qdefines partial charge of EP
Preparation input file for mdgx
Example of mdgx input file is listed below. Note that in case
of DCC, files with EP placement rules should be explicitly
specified in &files section
(see calc_energy_template+dcc.mdgx.in for details).
Listing 11: Example of mdgx input file (calc_energy_template.mdgx.in)
&files
-p COMPLEX_in.gaff.prmtop
-c COMPLEX_in.gaff.prmcrd
-o COMPLEX_out.mdgx.out
&end
&configs
verbose=1,
count=1,
maxcyc=1,
ncyc=1,
imin=2,
ntb=0,
cut=2000.0,
outbase ’COMPLEX_out.mdgx.out’,
write ’cdf’,
outsuff ’cdf’,
&end
Energy calculation
Finally, we calculate the free energy of a complex using
mdgx and compare them to the reference values.
Listing 12: Free energy calculation procedure (run_mdgx.sh)
# run mdgx on prepared input files
mkdir -p result/mdgx_out
cd result/mdgx_in
for i in H3N_mmff94 H3N_resp H3N_am1bcc H3N_dcc; do
for d in 01.6 01.8 02.0 02.5 03.0 03.5 04.0 04.5 05.0; do
mdgx -O -i "H2O_term_d_${d}+${i}.mdgx.in"
done
done
| Å | mmff94 | resp | am1bcc | dcc |
|---|---|---|---|---|
| 1.6 | -5.5098 | -5.6180 | -4.7096 | -10.2918 |
| 1.8 | -7.0008 | -7.0815 | -6.4034 | -10.1026 |
| 2.0 | -6.4261 | -6.4879 | -5.9692 | -8.5232 |
| 2.5 | -4.0321 | -4.0665 | -3.7774 | -4.9367 |
| 3.0 | -2.4671 | -2.4881 | -2.3114 | -2.9176 |
| 3.5 | -1.5747 | -1.5884 | -1.4729 | -1.8230 |
| 4.0 | -1.0486 | -1.0581 | -0.9786 | -1.1963 |
| 4.5 | -0.7222 | -0.7290 | -0.6720 | -0.8153 |
| 5.0 | -0.5099 | -0.5149 | -0.4727 | -0.5713 |